
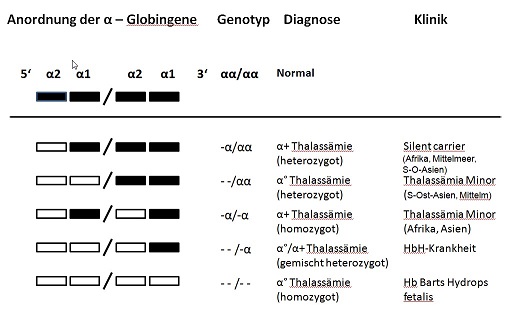
Es gibt 5 verschiedene alpha–Thalassämien. Wenn eines der 4 alpha–Gene fehlt macht sich das kaum bemerkbar (silent carrier) – es sind ja noch 3 aktive Gene übrig. Diese Form der alpha–Thalassämie nennt man „heterozygote alpha+ (alpha plus) Thalassämie“ oder Thalassämia Minima. Heterozygot bedeutet, dass man diese Gen-Veränderung nur von einem Elternteil geerbt hat. Das „Plus“ bzw. „Minima“ sagt, dass diese Genveränderung nicht schlimm ist. Wenn auf jedem Chromosom 16 ein α-Gen fehlt, nennt man diese Form homozygote alpha+ Thalassämie oder Thalassämia Minor, d.h. auf dem Chromosom 16 von Vater und Mutter fehlt je ein alpha–Gen. Dieses Kind hat statt 4 nur noch 2 alpha–Gene, aber auch das macht einen nicht krank: man hat fast immer einen normalen Hämoglobinwert, nur sind die roten Blutkörperchen deutlich kleiner als normal. In Afrika gibt es fast ausschließlich nur diese beiden Formen der alpha+-Thalassämie. Ca. 30% aller Menschen in Zentralafrika haben eine dieser beiden alpha-Thalassämie Formen, die nicht krankmachen.
Wenn beide alpha–Gene auf einem Chromosom 16 fehlen entsteht die heterozygote alpha0 Thalassämie, die zwar auch wie eine Thalassämia Minor (keine Anämie, aber kleine rote Blutkörperchen) aussieht, aber für die nächste Generation große Schwierigkeiten machen kann. Deshalb heißt diese Form der alpha–Thalassämie auch heterozygote alpha0 (alpha Null) Thalassämie. Wenn nämlich ein Kind von beiden Eltern eine alpha0-Thalassämie erbt und eine homozygote alpha0-Thalassämie hat, hat es einen sog. Hydrops fetalis: es kann überhaupt keine alpha-Ketten bilden, d.h. schon in der frühen Schwangerschaft hat das neue Baby (der Fet) eine schwere Anämie, entwickelt Wasseransammlungen im ganzen Körper (Hydrops) und stirbt in den ersten 4-5 Monaten der Schwangerschaft. Nicht nur das Kind ist gefährdet – auch die Mutter hat schwere Komplikationen in einer solchen Schwangerschaft. Die alpha0-Thalassämie gibt es vor allem in Süd-Ost-Asien, aber auch in der Türkei und im Mittleren Osten. In diesen Ländern gibt es aber auch die alpha+-Thalassämie.
Bei der Kombination einer heterozygoten alpha+ mit einer heterozygoten alpha0 Thalassämie (das betroffene Kind hat nur 1 statt 4 alpha-Gene) entsteht die Hämoglobin H – Krankheit. Wenn jemand nur 1 einziges alpha-Gen hat, werden nur sehr wenige alpha-Ketten gebildet und die vielen ß-Ketten, die keine Partner finden, tun sich als 4er Gruppe zusammen. Diese aus 4 ß-Ketten bestehenden Gebilde nennt man HbH. Sie können Sauerstoff binden, ihn aber nicht wieder abgeben und sie führen dazu, dass die roten Blutkörperchen, die sehr klein sind, vorzeitig in der Milz zerstört werden (Hämolyse). Die HbH-Krankheit geht mit einer mäßig starken Anämie einher, die Milz ist meist vergrößert aber man braucht, bis auf einige Ausnahmen, keine Transfusionen. Mit der Hämoglobin H Krankheit kann man ein weitgehend normales Leben führen. Die HbH-Krankheit gibt es in Süd-Ost Asien, aber auch in der Türkei und in den Ländern des Mittleren Ostens.
Bei allen Träger der alpha0-Thalassämie und allen, die eine HbH-Krankheit haben müssen die Partner auf alpha0-Thalassämie untersucht werden. Sind beide Eltern Träger der alpha0-Thalassämie, sollte Pränatale Diagnostik gemacht werden, um schon in der frühen Schwangerschaft herausfinden zu können, ob das Risiko eines Hydrops fetalis besteht. In einer solchen Situation sollte die Schwangerschaft so früh wie möglich abgebrochen werden.
